Zwei häufige genetische Mutationen, die das Rett-Syndrom auslösen, führen zu undichten Blutgefäßen im sich entwickelnden Gehirn. Das zeigt eine neue Studie des Picower Institute for Learning and Memory am MIT. Die Forscher führen das Problem auf eine Überexpression des microRNA-126-3p zurück und konnten zeigen, dass eine gezielte Absenkung dieser microRNA die Gefäßdefekte teilweise behebt. Die Ergebnisse erschienen in Molecular Psychiatry.
Das Rett-Syndrom ist eine schwere Entwicklungsstörung, die vor allem Mädchen betrifft und durch verschiedene Mutationen im MECP2-Gen verursacht wird. Die ersten Symptome treten meist im Alter von 2–3 Jahren auf – eine Phase, in der sich die Hirngefäße entscheidend entwickeln. Die MIT-Neuroforscher untersuchten daher, wie zwei häufige MECP2-Mutationen (R306C und R168X) die Gefäßentwicklung beeinträchtigen und zur neurologischen Symptomatik beitragen könnten.
Dafür entwickelten sie fortschrittliche humane Gewebekulturen aus induzierten pluripotenten Stammzellen (iPS-Zellen) von Rett-Patientinnen. Die endothelialen Zellen (Grundbausteine der Blutgefäße) wurden in einem Gel mit Fibroblasten kultiviert und über Mikrofluidik mit Kreislauf versehen. So entstanden dreidimensionale mikrovaskuläre Netzwerke, die mit und ohne Mutationen verglichen wurden.
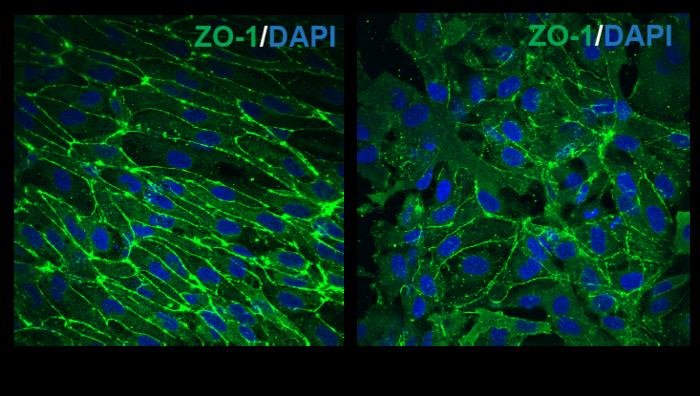
In den mutierten Kulturen zeigte sich eine reduzierte Expression und fehlerhafte Lokalisation des Proteins ZO-1, das für dichte Zell-Zell-Verbindungen in Gefäßen entscheidend ist. Die Gefäße waren dadurch deutlich durchlässiger als in den Kontrollkulturen. Auch in einem Modell der Blut-Hirn-Schranke (mit Astrozyten) traten Defekte auf. Neuronen, die dem Medium aus Rett-Gefäßen ausgesetzt waren, zeigten verringerte elektrische Aktivität – ein Hinweis darauf, dass undichte Gefäße die neuronale Funktion stören könnten.
Die Wissenschaftler fanden heraus, dass MECP2-Mutationen zu einer Überexpression von miRNA-126-3p führen, die wiederum ZO-1 und weitere für die Gefäßintegrität wichtige Moleküle herunterreguliert. Durch Behandlung mit einem Antisense-Molekül, das miRNA-126-3p absenkt, konnte die ZO-1-Expression gesteigert und die Barrierefunktion teilweise wiederhergestellt werden.
„Eine Rolle von microRNAs beim Rett-Syndrom war bekannt, aber dass miRNA-126-3p direkt downstream von MECP2 liegt und die endotheliale Dysfunktion verursacht, ist ein wichtiges Puzzlestück“, sagte Seniorautor Mriganka Sur, Newton Professor of Neuroscience am MIT.
Die Ergebnisse deuten darauf hin, dass Gefäßprobleme eine zentrale Rolle in der Pathologie des Rett-Syndroms spielen könnten. Ein Medikament, das miR-126 hemmt (miRisten), wird derzeit bei Leukämie klinisch getestet. Die Forscher planen, es in Rett-Mausmodellen zu prüfen.
Die Studie entstand in Zusammenarbeit mit dem Labor von Roger D. Kamm (MIT) und wurde unter anderem von den National Institutes of Health, der Freedom Together Foundation und dem Simons Center for the Social Brain finanziert.
Molecular Psychiatry
DOI
Bildnachweis
Tatsuya Osaki/MIT Picower Institute